

Covalent Peptide Evolution: Redefining Protein–Protein Interaction Inhibition Through Phage Display.

Nelson Santiago Vispo 1*
1*Clinical Biotec SL. and Bionatura Journal. Madrid. 28029. Spain.
*Correspondence: santiago@clinicalbiotec.com
ABSTRACT
Covalent cyclic
peptides represent a transformative approach for targeting challenging
protein-protein interactions (PPIs) characterized by flat, extensive binding
surfaces. Recent advances in electrophilic phage display now enable the
evolution of these peptides through integrating sulfur(VI) fluoride exchange
(SuFEx) chemistry with functional selection strategies. This innovative
platform combines genetic encoding with site-specific cyclization and warhead
incorporation to generate high-affinity, irreversible binders. When targeting
the SARS-CoV-2 Spike-ACE2 interface, the approach produced sub-100 nM
inhibitors with >10-fold improved potency over non-covalent analogues. The
methodology's success against this clinically relevant target underscores its
potential to address longstanding challenges in PPI modulation, particularly
for high-value targets in oncology and neurodegeneration. By combining covalent
engagement with phage display's evolutionary power, this technology establishes
a new paradigm for developing mechanistically validated peptide therapeutics
against previously intractable interactions.
Keywords: covalent peptide inhibitors, cyclic
peptide therapeutics, phage display evolution, protein-protein interaction
inhibition, SuFEx chemistry, irreversible binders, SARS-CoV-2 inhibitors,
Spike-ACE2 disruption, electrophilic warheads, undruggable targets, functional
selection, peptide macrocycles, PPI drug discovery, covalent phage display
INTRODUCTION
Targeting protein-protein interactions (PPIs)
remains a major challenge in drug discovery, largely due to their broad and
topologically flat interfaces, which are poorly suited to conventional
small-molecule inhibitors.¹ Thanks to their conformational
stability and selective binding, cyclic peptides are increasingly recognized as
valuable tools for modulating PPIs.² For example, the c-Myc/Max interaction—a
pivotal oncology target with a flat, extended interface—has resisted decades of
small-molecule discovery efforts.¹ However, transitioning from reversible to
covalent binding—advantageous for its prolonged engagement and
selectivity—remains constrained by the incompatibility of electrophilic
chemistry with standard genetic encoding systems.⁵
Recent advances by Wang et al. ⁶,
underscored in a commentary by Jin⁷, introduce a novel approach to phage
display that incorporates electrophilic elements to evolve covalent peptide
binders.
A
Dual-Layered Evolutionary Strategy
Wang et al. engineered phage-displayed peptide
libraries incorporating dibromoaryl fluorosulfate linkers,⁶ introducing
sulfur(VI) fluoride exchange (SuFEx) electrophiles that react selectively with
nucleophilic amino acid residues (Tyr, Lys, His) near PPI interfaces. The
AXCX₇CG format enables two cysteines for cyclization via dibromoaryl
fluorosulfate linkers while installing a SuFEx warhead—a sulfur(VI)-based
electrophile that reacts selectively under physiological conditions.⁶
The peptides were genetically encoded and fused
to the N-terminus of the filamentous phage protein pIII. They were chemically
cyclized in situ upon phage assembly, yielding a covalent cyclic peptide–phage
library.
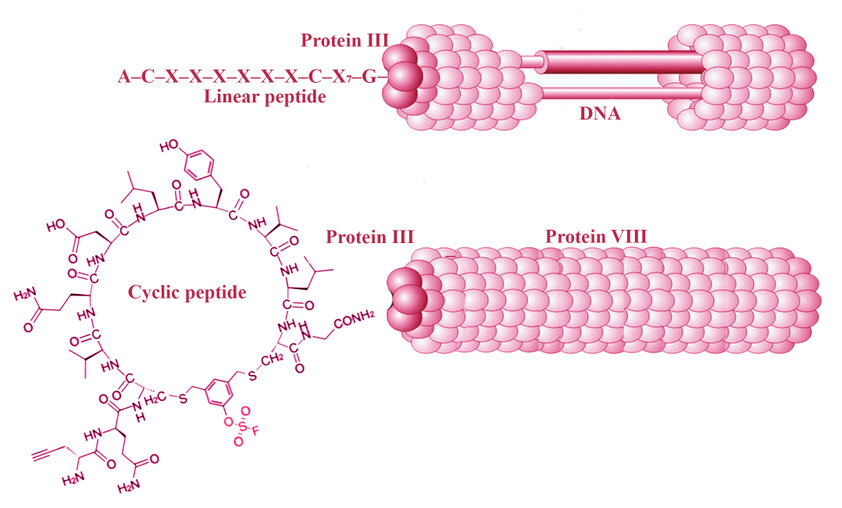
Figure 1. Workflow for Covalent Cyclic Peptide Evolution via Electrophilic Phage Display. Schematic representation of the library generation and selection process described by Wang et al.⁶ Peptides with the sequence AXCX₇CG are genetically encoded and fused to the N-terminus of the filamentous phage protein pIII. The linear peptides are displayed on the phage surface and contain two cysteine residues, enabling chemical cyclization via a dibromoaryl fluorosulfate linker. This linker forms a thioether bridge between the cysteines and installs a SuFEx electrophilic warhead (red). The resulting covalent cyclic peptide-phage library undergoes iterative rounds of (1) positive selection on immobilized target protein (e.g., SARS-CoV-2 Spike), followed by (2) functional counterselection with a competitor (e.g., ACE2 or a covalent nanobody), To enrich for high-affinity, functional binders. Selected clones are sequenced and validated for covalent target engagement and biological activity. [Adapted from Wang et al. 6 and Jin et al. 2025 7]
Applied to the SARS-CoV-2 Spike–ACE2
interaction, the platform employed three rounds of positive selection followed
by two rounds of functional counterselection using ACE2 and a covalent nanobody
competitor. This ensured the enrichment of peptides binding specifically at the
functional interface. The lead peptide exhibited irreversible binding and an
IC₅₀ of 72 nM against the Omicron BA.2 variant, representing a >10-fold
improvement compared to non-covalent peptide inhibitors of Spike–ACE2, such as
the linear mini protein 2P2 (IC₅₀ ≈ 700 nM).¹⁵
Beyond
Affinity: A Function-First Paradigm
The innovation lies not just in chemistry but
in evolutionary logic. Implementing counterselection against non-functional
binders shifts the strategy from passive affinity enrichment to mechanistically
driven selection. This mirrors Wang et al.'s counterselection strategy, where
functional pressure (ACE2 competition) was critical to isolate
interface-binding peptides—only 0.1% of initial hits passed this stringent
filter.⁶
The framework builds on prior work with cyclic
peptide libraries for therapeutic applications, including our efforts using
proapoptotic cyclic peptides to impair CK2-mediated phosphorylation and induce
antitumor effects.⁴ Future developments might include:
- In silico electrophile pre-screening to optimize chemical reactivity⁵,⁹
- Modular warhead libraries to expand target scope⁵,⁶
- Orthogonal counterselection against off-targets or homologous proteins⁶,⁷
For example, counterselection against BRD4 BD1
could yield inhibitors selective over BD2, addressing a key challenge in BET
protein targeting.⁸,¹²
Structural
and Therapeutic Advantages of Cyclic Peptides
Conformational
Rigidity: Cyclization reduces the entropic penalty of
binding by ~5 kcal/mol compared to linear analogs.³
Proteolytic Stability: Half-lives exceed 24 hours in serum vs. <1 hour for linear versions.¹¹
Permeability: Despite chemical engineering advances, only ~20% of cyclic peptides achieve intracellular concentrations >1 µM without modifications such as N-methylation or stapling.¹²
Drug-like Properties: Cyclic peptides combine the specificity of biologics with small-molecule permeability, enabling them to modulate previously intractable PPIs.¹⁰, ¹²
Proteolytic Stability: Half-lives exceed 24 hours in serum vs. <1 hour for linear versions.¹¹
Permeability: Despite chemical engineering advances, only ~20% of cyclic peptides achieve intracellular concentrations >1 µM without modifications such as N-methylation or stapling.¹²
Drug-like Properties: Cyclic peptides combine the specificity of biologics with small-molecule permeability, enabling them to modulate previously intractable PPIs.¹⁰, ¹²
Therapeutic
Horizons
This technology opens new doors to previously "undruggable"
targets:
- In oncology, covalent inhibition of c-Myc could address its "undruggable" status in over 70% of human cancers.
- In neurodegeneration, targeting Tau's oligomerization interfaces may help block early aggregation events in Alzheimer's disease.¹,¹²
Its applications extend across oncology,
virology, and neurodegenerative disease and support the development of chemical
biology tools for dissecting transient or disordered PPIs.⁶,7,¹²
CONCLUSIONS
The strategy presented by Wang et al. represents
a significant evolution in covalent ligand engineering, merging chemical
reactivity with selective evolutionary pressure within the phage display
framework. By
embedding electrophilic chemistry and functional selection pressure into phage
display, this approach offers a powerful platform to generate mechanistically
active covalent cyclic peptides, poised to unlock the therapeutic potential of
PPIs long considered intractable.
In addition to its therapeutic potential, this
methodology may facilitate the development of specialized chemical probes,
aiding the validation of complex protein interaction targets in biomedical
research.
Ethics statement (even if not applicable):
This article contains no studies with human or animal subjects performed by the author.
This article contains no studies with human or animal subjects performed by the author.
Conflict of interest:
N.S.V. is affiliated with 1st Clinical Biotec SL. The author declares no other competing interests.
N.S.V. is affiliated with 1st Clinical Biotec SL. The author declares no other competing interests.
REFERENCES
1.
Scott DE, Bayly AR, Abell C, Skidmore J. Small
molecules, big targets: drug discovery faces the protein–protein interaction
challenge. Nat Rev Drug Discov. 2016;15(8):533–550.
2.
González-Muñiz R, Bonache M, De Vega M.
Modulating protein–protein interactions by cyclic and macrocyclic peptides.
Molecules. 2021;26(2):445.
3.
Cardote T, Ciulli A. Cyclic and macrocyclic
peptides as chemical tools to recognize protein surfaces and probe
protein–protein interactions. ChemMedChem. 2016;11(8):787–794.
4.
Perea SE, Reyes O, Puchades Y, Mendoza O, Vispo
NS, Torrens I, et al. Antitumor effect of a novel proapoptotic peptide that
impairs phosphorylation by protein kinase 2. Cancer Res. 2004;64(19):7127–7129.
5.
Hampton JT, Liu WR. Expanding the genetic code
for covalent peptide evolution. Chem Rev. 2024;124(11):6051–6077.
6.
Wang S, Zhang Y, Li J, et al. Covalent peptide
inhibitors from electrophilic phage display target Spike–ACE2 interaction. J Am
Chem Soc. 2025;147(15):7461–7475.
7.
Jin G. Fishing for covalent peptides. Nat Chem
Biol. 2025;21(5):456–462.
8.
Chen S, Wang XS, Zheng M. Next-generation phage
display for covalent inhibitor discovery. Nat Biotechnol. 2021;39(4):490–498.
9.
Ekanayake AI, Chen L, Yan K, et al. Covalent
peptide ligands from electrophilic phage display libraries. J Am Chem Soc.
2021;143(12):5497–5507.
10. Cheng J,
Zhou J, Kong L, et al. Stabilized cyclic peptides as modulators of
protein–protein interactions: promising strategies and biological evaluation.
RSC Med Chem. 2023;14(12):1204–1221.
11. Qian Z,
Dougherty PG, Pei D. Targeting intracellular protein–protein interactions with
cell-permeable cyclic peptides. Curr Opin Chem Biol. 2017;38:80–86.
12. Buyanova
M, Pei D. Targeting intracellular protein–protein interactions with macrocyclic
peptides. Trends Pharmacol Sci. 2021;42(1):17–29.
13. Kosugi
T, Ohue M. Design of cyclic peptides targeting protein–protein interactions
using AlphaFold. Int J Mol Sci. 2023;24(19):14100.
14. Cao L,
Goreshnik I, Coventry B, et al. De novo design of picomolar SARS-CoV-2
miniprotein inhibitors. Science. 2020;370(6515):426–431.
15. McHugh
S, Rogers J, Solomon S, et al. Computational methods to design cyclic peptides.
Curr Opin Chem Biol. 2016;34:70–77.
16. Hashemi
Z, Zarei M, Fath M, et al. In silico approaches for the design and optimization
of interfering peptides against protein–protein interactions. Front Mol Biosci.
2021;8:669431.
Received: April 20, 2025 / Accepted:
May 2, 2025 / Published: June 15, 2025
Citation: Santiago Vispo N. Covalent Peptide Evolution: Redefining
Protein–Protein Interaction Inhibition Through Phage Display. Bionatura
Journal. 2025;2(2):16. doi: 10.70099/BJ/2025.02.02.16
Additional
information Correspondence
should be addressed to santiago@clinicalbiotec.com
Peer review information. Bionatura thanks anonymous reviewer(s) for their
contribution to the peer review of this work using https://reviewerlocator.webofscience.com/
ISSN.3020-7886
All articles published by Bionatura Journal
are made freely and permanently accessible online immediately upon publication,
without subscription charges or registration barriers.
Publisher's Note: Bionatura Journal stays neutral concerning
jurisdictional claims in published maps and institutional affiliations.
Copyright: © 2025 by the authors. They were submitted for possible open-access
publication under the terms and conditions of the Creative Commons Attribution
(CC BY) license (https://creativecommons.org/licenses/by/4.0/).